|
OpenMS
2.4.0
|
|
OpenMS
2.4.0
|
The following sections explain the example pipelines TOPPAS comes with. You can open them by selecting File > Open example file. All input files and parameters are already specified, so you can just hit Pipeline > Run (or press F5) and see what happens.
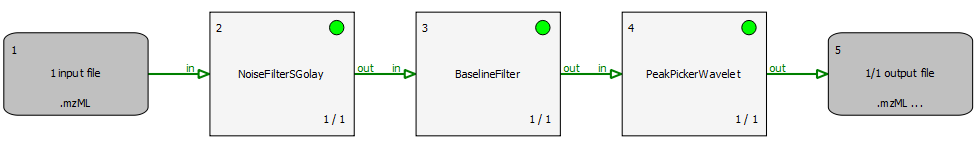
The file peakpicker_tutorial.toppas contains a simple pipeline representing a common use case: starting with profile data, the noise is eliminated and the baseline is subtracted. Then, PeakPickerWavelet is used to find all peaks in the noise-filtered and baseline-reduced profile data. This workflow is also described in the section Profile data processing. The individual steps are explained in more detail in the TOPPView tutorial: Smoothing raw data, Subtracting a baseline from a spectrum, and Picking peaks.
This section describes an example identification pipeline contained in the example directory, Ecoli_Identification.toppas. It is shipped together with a reduced example mzML file containing 139 MS2 spectra from an E. coli run on an Orbitrap instrument as well as an E. coli target-decoy database.
We use the search engine OMSSA (Geer et al., 2004) for peptide identification. Therefore, OMSSA must be installed and the path to the OMSSA executable (omssacl) must be set in the parameters of the OMSSAAdapter node.
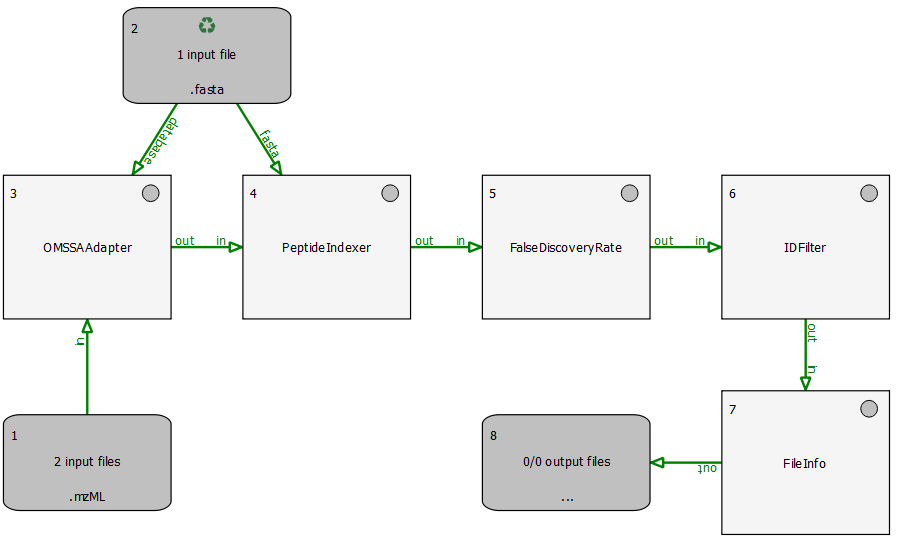
Extensions to this pipeline would be to do the annotation of the spectra with multiple search engines and combine the results afterwards, using the ConsensusID TOPP tool.
The results may be exported using the TextExporter tool, for further analysis with different tools.
The simple pipeline described in this section (BSA_Quantitation.toppas) can be used to quantify peptides that occur on different runs. The example dataset contains three different bovine serum albumin (BSA) runs. First, FeatureFinderCentroided is called since the dataset is centroided. The results of the feature finding are then annotated with (existing) identification results. For convenience, we provide these search results from OMSSA with peptides with an FDR of 5% in the BSA directory.
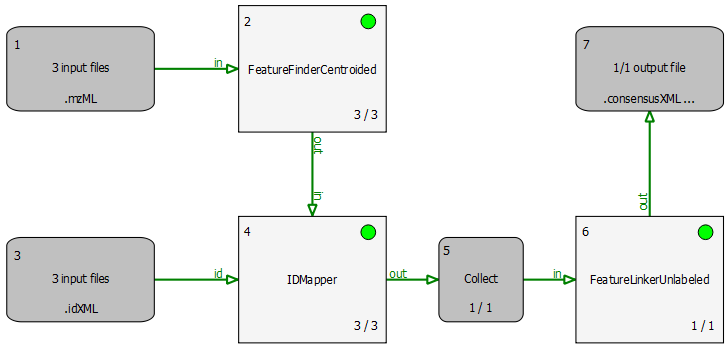
Identifications are mapped to features by the IDMapper. The last step is performed by FeatureLinkerUnlabeled which links corresponding features. The results can be used to calculate ratios, for example. The data could also be exported to a text based format using the TextExporter for further processing (e.g., in Microsoft Excel).
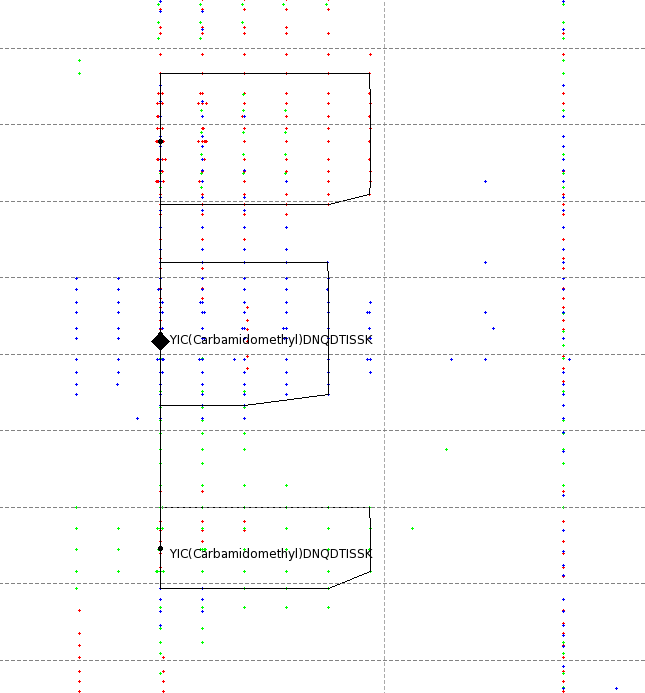
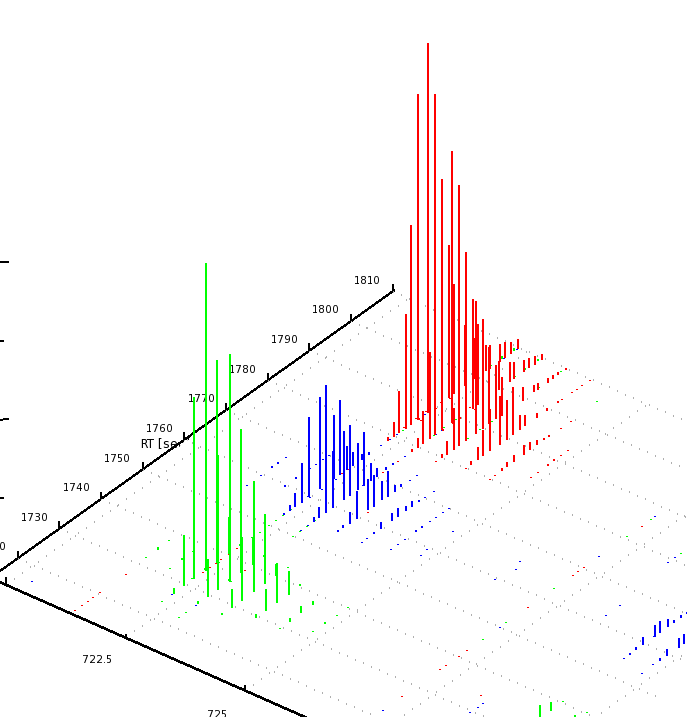
The results can be opened in TOPPView. The next figures show the results in 2D and 3D view, together with the feature intermediate results. One can see that the intensities and retention times are slightly different between the runs. To correct for retention times shift, a map alignment could be done, either on the spectral data or on the feature data.
The following example is actually not a useful workflow but is supposed to demonstrate how merger and collector nodes can be used in a pipeline. Have a look at merger_tutorial.toppas:
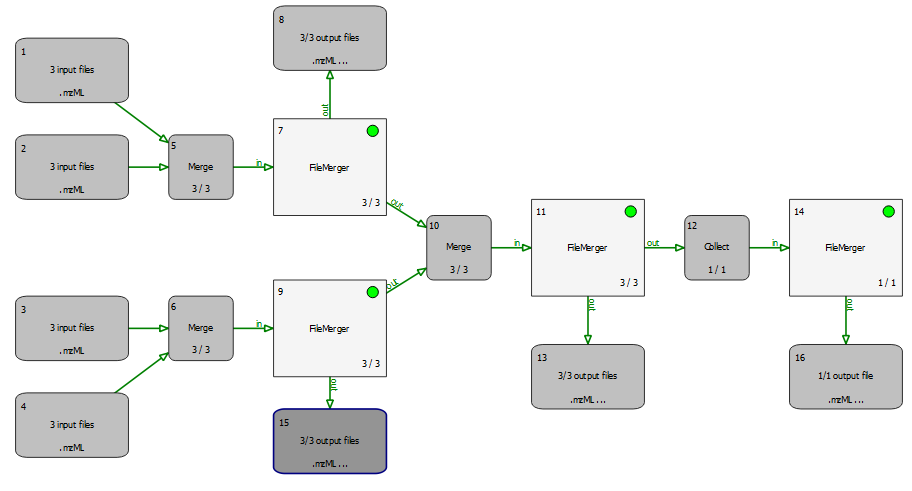
As its name suggests, a merger merges its incoming file lists, i.e., files of all incoming edges are appended into new lists (which have as many elements as the merger has incoming connections). All tools this merger has outgoing connections to are called with these merged lists as input files. All incoming connections should pass the same number of files (unless the corresponding preceding tool is in recycling mode).
A collector node, on the other hand, waits for all rounds to finish before concatenating all files from all incoming connections into one single list. It then calls the next tool with this list of files as input. This will happen exactly once during the entire pipeline run.
In order to track what is happening, you can just open the example file and run it. When the pipeline execution has finished, have a look at all input and output files (e.g., select Open in TOPPView in the context menu of the input/output nodes). The input files are named rt_1.mzML, rt_2.mzML, ... and each contains a single spectrum with RT as indicated by the filename, so you can easily see which files have been merged together.
 1.8.14
1.8.14