|
OpenMS
2.6.0
|
The goal of map alignment is to transform different HPLC-MS maps (or derived maps) to a common retention time axis. It corrects for shifted and scaled retention times, which may result from changes of the chromatography.
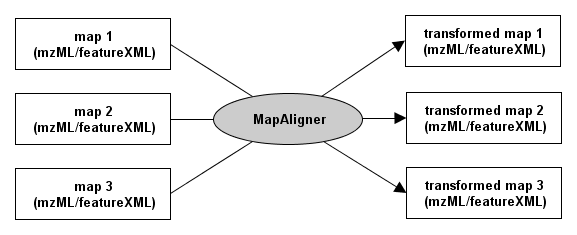
The different MapAligner tools take n input maps, de-warp them and store the n de-warped maps. The following image shows the general procedure:
There are different map alignment tools available. The following table gives a rough overview of them:
| Application: MapAlignerPoseClustering Applicable to: feature maps, peak maps |
| Description: This algorithm does a star-wise alignment of the input data. The center of the star is the map with most data points. All other maps are then aligned to the center map by estimating a linear transformation (shift and scaling) of retention times. The transformation is estimated using a pose clustering approach as described in doi:10.1093/bioinformatics/btm209 |
| Application: MapAlignerIdentification Applicable to: feature maps, consensus maps, identifications |
| Description: This algorithm utilizes peptide identifications, and is thus applicable to files containing peptide IDs (idXML, annotated featureXML/consensusXML). It finds peptide sequences that different input files have in common and uses them as points of correspondence. From the retention times of these peptides, transformations are computed that convert each file to a consensus time scale. |
| Application: MapAlignerSpectrum Applicable to: peak maps |
| Description: This experimental algorithm uses a dynamic-programming approach based on spectrum similarity for the alignment. The resulting retention time mapping of dynamic-programming is then smoothed by fitting a spline to the retention time pairs. |
| Application: MapRTTransformer Applicable to: peak maps, feature maps, consensus maps, identifications |
| Description: This algorithm merely applies a set of transformations that are read from files (in TransformationXML format). These transformations might have been generated by a previous invocation of a MapAligner tool. For example, you might compute a transformation based on identifications and then apply it to the features or raw data. The transformation file format is not very complicated, so it is relatively easy to write (or generate) your own transformation files. |
 1.8.16
1.8.16

