|
OpenMS
2.7.0
|
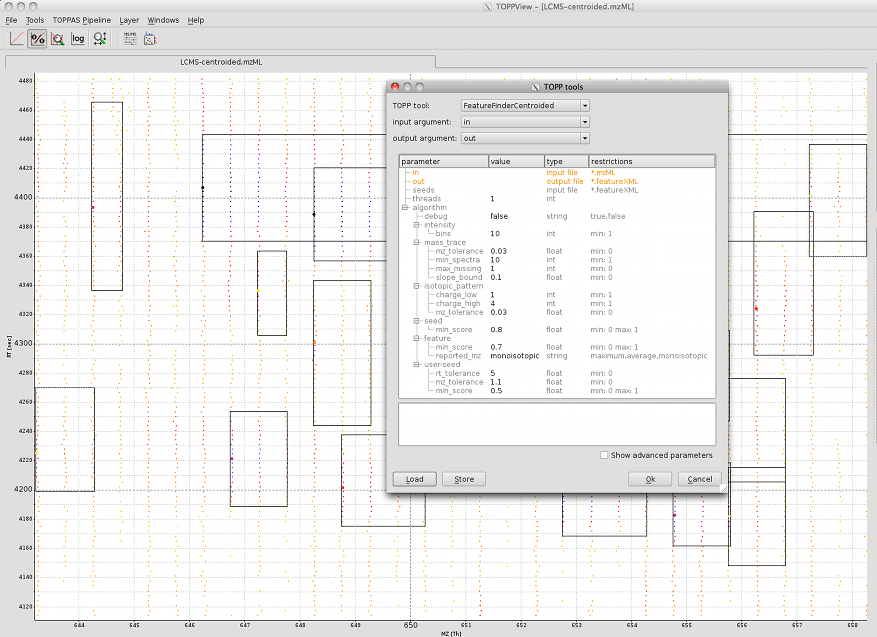
To quantify peptide features, TOPP offers the FeatureFinder tools. In this section the FeatureFinderCentroided is used, which works on centroided data only. There are other FeatureFinders available that also work on profile data.
For this example the file LCMS-centroided.mzML from the examples data is used (File > Open example data). In order to adapt the algorithm to our data, some parameters have to be set.
Intensity
The algorithm estimates the significance of peak intensities in a local environment. Therefore, the HPLC-MS map is divided into n times n regions. Set the intensity:bins parameter to 10 if you are working on a whole map. If you are working on a small region only, you should set it to 1.
Mass trace
For the mass traces, you have to define the number of adjacent spectra in which a mass has to occur (mass_trace:min_spectra). In order to compensate for peak picking errors, missing peaks can be allowed (mass_trace:max_missing) and a tolerated mass deviation must be set (mass_trace:mz_tolerance).
Isotope pattern
The expected isotopic intensity pattern is estimated from an averagene amino acid composition. The algorithm searches all charge states in a defined range (isotopic_pattern:change_min to isotopic_pattern:change_max). Just as for mass traces, a tolerated mass deviation between isotopic peaks has to be set (isotopic_pattern:mz_tolerance).
The image shows the centroided peak data and the found peptide features. The used parameters can be found in the TOPP tools dialog.
 1.9.1
1.9.1

