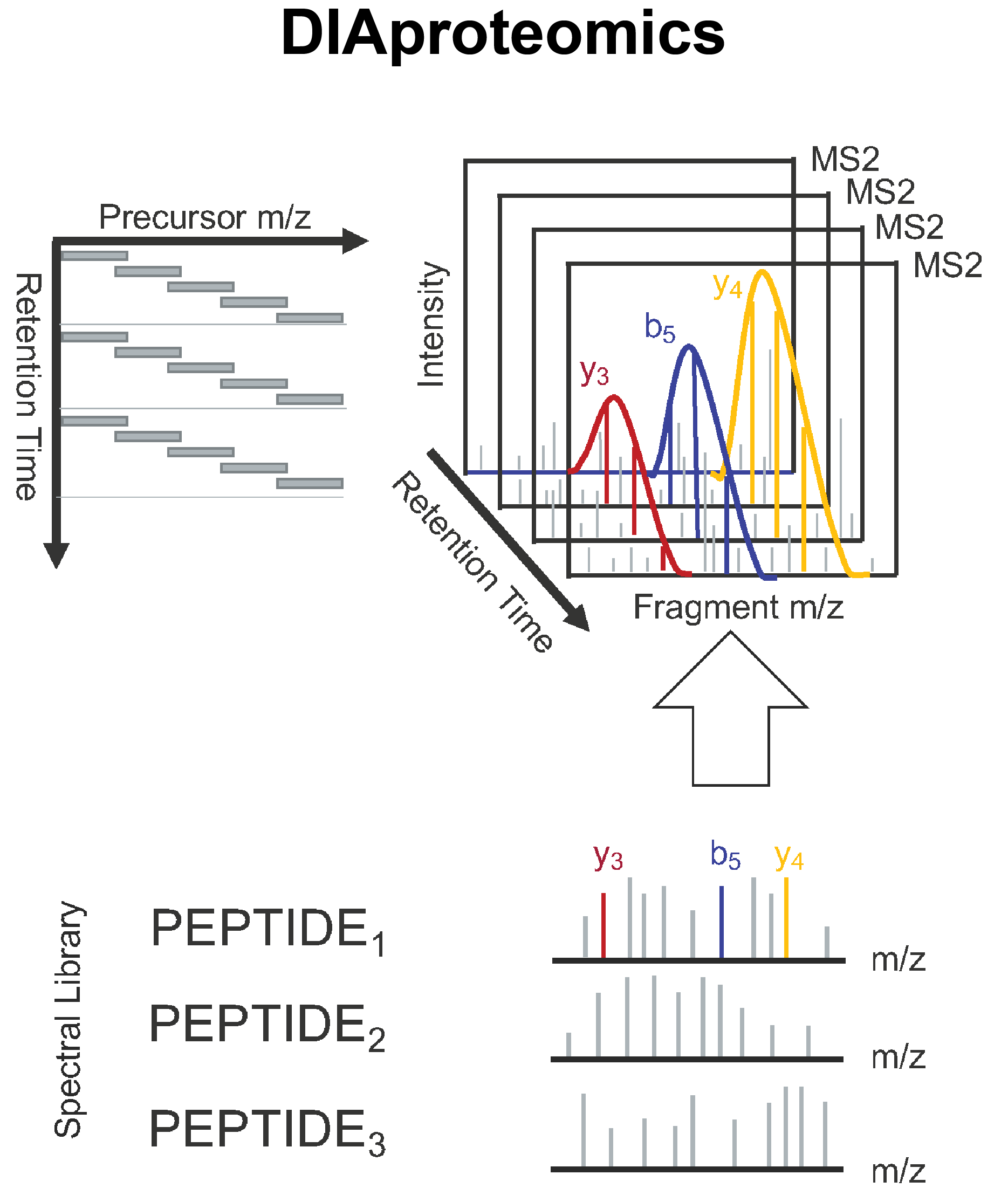
DIAproteomics is a bioinformatics analysis pipeline used for quantitative processing of data independent (DIA) proteomics data.
The workflow is based on the OpenSwathWorkflow for SWATH-MS proteomic data. DIA RAW files (mzML) serve as inputs and library search is performed based on a given input spectral library. If specified internal retention time standards (irts) will be used to align library and DIA measurements into the same retention time space. FDR rescoring is applied using Pyprophet based on a competitive target-decoy approach on peakgroup or global peptide and protein level. Optionally EasyPQP can be used for library generation and DIAlignR for chromatogram alignment and quantification.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker / singularity containers making installation trivial and results highly reproducible.
The Nextflow Workflow is available at:
https://github.com/nf-core/diaproteomics
See also the detailed description of parameters and workflow output at: