OpenPepXL: an open source peptide cross-link identification tool
OpenPepXL is a protein-protein cross-link identification tool implemented in C++ as part of OpenMS. It works with all uncleavable labeled and label-free cross-linkers but not (yet) with cleavable ones.
Requirements
The current version of OpenPepXL, version 1.1 is available as part of OpenMS 2.5. Installers for Windows, MacOS and Linux can be found here. OpenPepXL can be used effectively on a desktop computer with 16BG of memory.
Input and output
As input OpenPepXL requires
- mzML file(s) with centroided MS2 spectra (centroided either on acquisition, on conversion to mzML or using the OpenMS tool PeakPickerHiRes)
- fasta file with the protein database
As output OpenPepXL supports
- idXML format, which is OpenMS’ internal format compatible with various tools in OpenMS, e.g. for quantification and visualization
- mzIdentML 1.2 format (Vizcaíno et al., 2017) will allow complete submission of cross-linking data to the PRIDE database and ProteomeXchange (Ternent et al., 2014)
- xQuest format (Rinner et al., 2008) and thus compatible with xProphet (Leitner et al., 2014) for FDR estimation and xTract (Walzthoeni et al., 2015) for quantification, as well as the UCSF Chimera plug-in Xlink Analyzer (Kosinski et al., 2015) for visualizing and analyzing cross-links on structures.
Beginners: use OpenPepXL through the workflow manager KNIME
To use OpenPepXL in KNIME, first install the newest KNIME release and install OpenMS 2.5 through the KNIME community contributions. Instructions are at https://openms.readthedocs.io/en/latest/getting-started/knime-get-started.html).
For a general introduction to KNIME and OpenMS start here: https://openms.readthedocs.io/en/latest/getting-started/knime-get-started.html Building workflows is done by finding tools in the tool list (e.g. by text search) and dragging and dropping their nodes on the workflow pane. The output from one node can be connected to the input of another node by dragging the mouse from one port to another. The ports are the rectangular or triangular symbols on the left and right sides of tool nodes. Inputs are always on the left, outputs on the right. Each node can be configured by double-clicking on its icon or right-clicking and selecting the configure option. A pop-up window will then list the parameters of the node. The figures below show example workflows for OpenPepXL (labeled cross-linkers) and OpenPepXLLF (unlabeled cross-linkers).
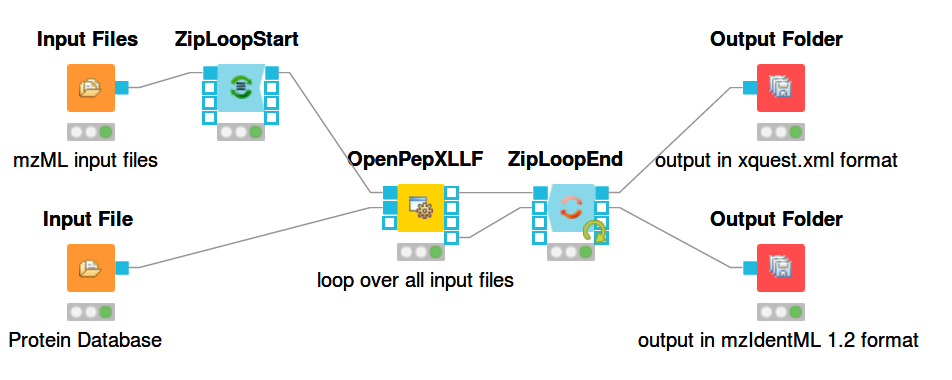
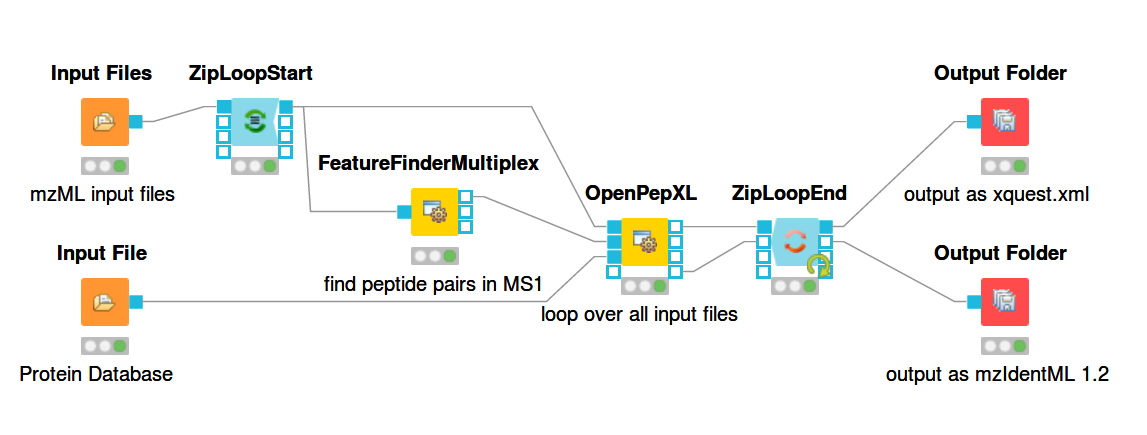
For reading in input files there are the nodes Input File and Input Files. Input File has one file path as its parameter and can be used for single files, like a protein database. The Input Files node takes a list of file paths for other nodes to iterate over. The ZipLoopStart and ZipLoopEnd nodes allow for a section of the workflow to iterate over multiple entries in a list. In this case FeatureFinderMultiplex and OpenPepXL or OpenPepXLLF are run multiple times, once for each input file defined in the Input Files node. The Output Folder nodes have a path to a directory as their parameter and write out all incoming data as files into that directory. The names of these files will be equal to the names of the mzML input files, but with a different file ending. The ZipLoopEnd node collects all the results of the loop before sending them further to the output nodes, so the results are written once after all processing is finished. Putting an Output Folder node inside the loop, e.g. right after OpenPepXL, will write out the results of the current input file right after OpenPepXL is finished with it. This type of workflow will go through the input files iteratively one by one and parallelization for each run can be set using the -threads parameter of OpenPepXL. To run a KNIME workflow, click on the green arrow button above the workflow pane.
Below you can see a more complex workflow with FDR estimation and additional filtering. This is a label-free OpenPepXL workflow available for download at the OpenMS 2.5 page.
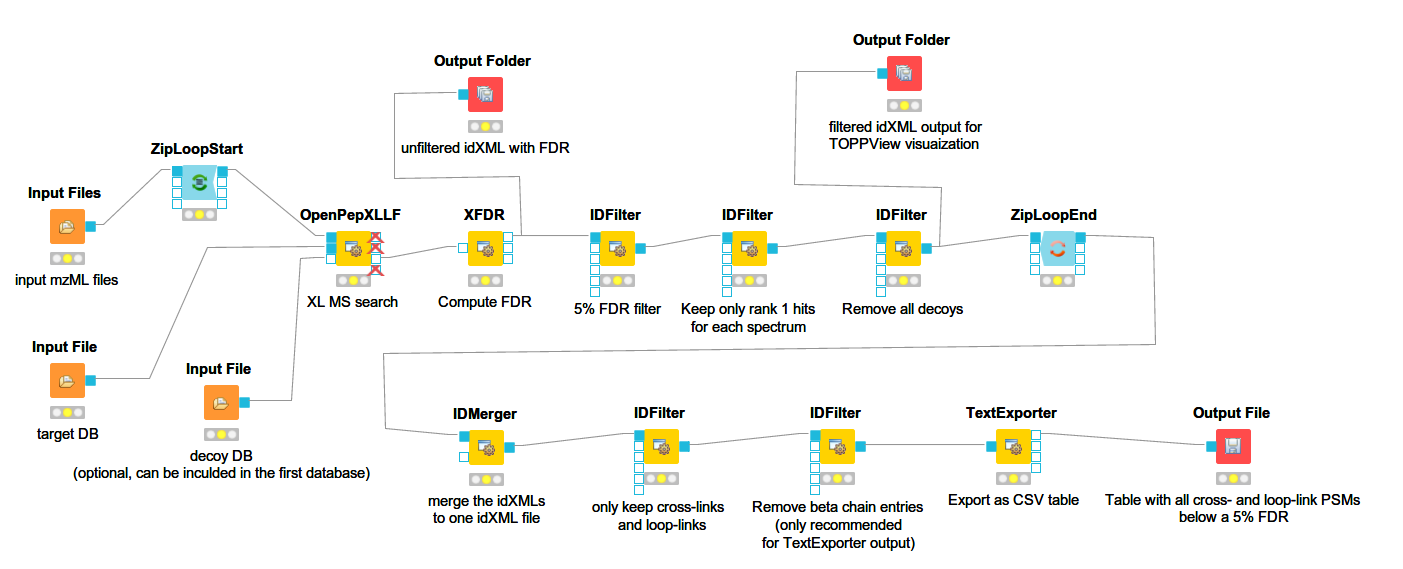
It will run OpenPepXLLF on all mzML input files, run the FDR estimation, filter by 5% FDR, remove decoys and write out idXML files for visualization in TOPPView (more below), as well as an CSV file containing all 1st ranked PSMs with FDR < 5% from all input mzML files. To run FeatureFinderMultiplex and OpenPepXL for labeled cross-link search, remove the OpenPepXLLF node and drag those two in. Connect the inputs and outputs in a similar way to the workflow above. You can also add a PeakPickerHiRes node for peak picking in front of FeatureFinderMultiplex or OpenPepXLLF if needed and adapt the filters as needed. The IDFilter node has a lot of options, but the only one used here is to filter by specific meta values of each ID (cross-link). The parameter -remove_peptide_hits_by_metavalue accepts three text strings that make up a condition. All IDs that fulfill this condition are accepted, others are filtered out. Look at the conditions set in the nodes of the workflow to get an idea about how this works. Most of the columns in the table returned by TextExporter can be used for filtering.
If you have any questions, suggestions, or bug reports, please visit the support page, open an issue on github, or write an email to eugen.netz@tuebingen.mpg.de
Advanced: use OpenPepXL as stand-alone application
The current stand-alone version of OpenPepXL, version 1.1 is available as part of OpenMS 2.5. Installers for Windows, MacOS and Linux can be found here.
Introduction to tool settings for command line use
All the TOPP (The OpenMS Proteomics Pipeline) tools are controlled through so-called INI files. To generate a tool specific INI file with default settings, call the tool executable with the parameter -write_ini filename.ini
OpenPepXLLF -write_ini OPXL.ini
To edit the settings open the .ini file in the TOPP tool INIFileEditor. An example of an INI file is shown in the figure.
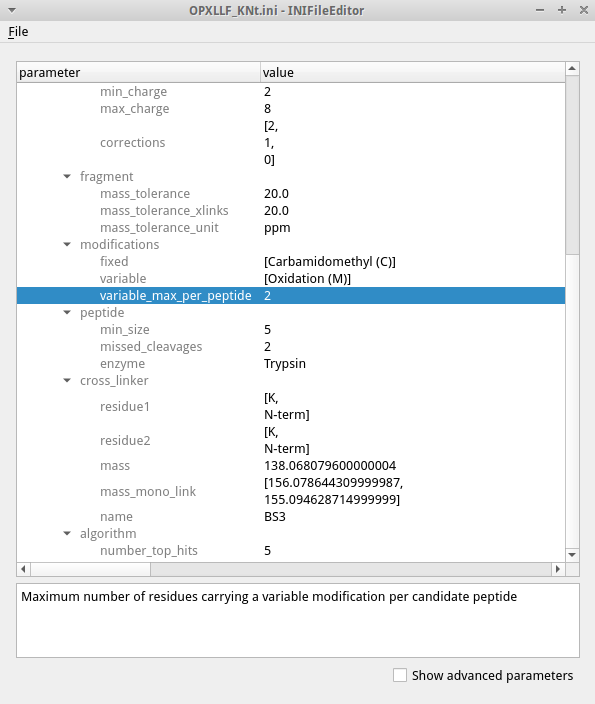
The INIFileEditor shows a description of each parameter at the bottom and helps to fill out many parameters, e.g. by using a file browser to select input and output files and showing the possible choices of parameters with limited options. To see the full list of parameters, including advanced parameters that should not be necessary for most users, check the box for advanced parameters on the bottom. The INI files can also be edited using a text editor when opening a GUI is not possible, e.g. when working on a remote server.
To run a tool using the edited INI file, call the tool executable with the parameter -ini filename.ini.
Example:
OpenPepXLLF -ini path/to/OPXL.ini
One INI file can be used for several runs with different parameters (e.g. another input and output file) by explicitly giving the tool additional parameters on the command line. These command line parameters will have higher priority than the values for these parameters in the INI file. This allows to have a fixed set of parameters using an INI file with a subset of variable parameters that you can change for each run. Examples:
OpenPepXLLF -ini path/OPXL.ini -in path/input_file_01.mzML -out_idXML path/output_file_01.idXML
OpenPepXLLF -ini path/OPXL.ini -in path/input_file_02.mzML -out_idXML path/output_file_02.idXML
All parameters adjustable through an INI file are also adjustable through the command line. Many parameters are grouped into categories, e.g. all OpenPepXL parameters concerning precursor masses are in the precursor group. To adjust these through the command line, you have to use the group name as a prefix, e.g. -precursor:mass_tolerance 10 and -precursor:mass_tolerance_unit ppm. The examples shown here assume OpenMS with OpenPepXL is installed on a Linux computer and the binaries are in the PATH. On Windows computers the binaries of the tools would end with .exe, otherwise everything should work in the same way.
Setting up and running OpenPepXLLF (label-free linkers):
-
Generate an INI file for OpenPepXLLF (
OpenPepXLLF -write_ini OPXL.ini). Open the generated INI file with the INIFileEditor. Choose your mzML input file (parameter: -in) and output files in any of the three supported formats (-out_xquestxml, -out_idxml, -out_mzid). If you do not specify any of these, the tool will run through to the end, but will not write out any results! Note that you can use multiple output formats. -
The preferred output format is the internal identification format of OpenMS: idXML. This is most compatible with XFDR and other post-processing and filtering tools within OpenMS, like IDFilter and IDMerger. Results can be formatted into other formats than idXML, e.g. mzIdentML using IDFileConverter or into a CSV table using TextExporter. Before running the tool, make sure you set up the protein database in a way that is compatible with XFDR (see the next section).
-
With the parameter -threads you can choose the number of CPU threads the tool will use for this run with the given input file. Alternatively you can start the tool with different parameters or input files in parallel using multiple terminals. Processing multiple files in parallel will be a bit more time efficient, but will add up the required memory for each run. Using the -threads parameter to process one input file in parallel will use the same amount of memory as processing it on a single thread.
-
Adapt the precursor and fragment mass tolerances to your MS instrument (for Orbitrap data, usually a precursor mass tolerance of 10 ppm and a fragment mass tolerance of 20 ppm) and add fixed and variable modifications, which you expect in your samples (aside from the cross-linker).
-
In the cross-linker category you can define your cross-linker. The default settings are for DSS and are also correct for BS3. The -residue1 and -residue2 parameters accept lists of residues for each reactive group of the linker, so that you can define any heterobifunctional cross-linker. N-term and C-term are also valid entries for these two parameters and will link protein termini.
-
Run the tool using the command line:
OpenPepXLLF -ini OPXL.ini
or in case of multiple input files and/or parameter sets
OpenPepXLLF -ini OPXL1.ini -in path/file1.mzML -out_idXML path/file1.idXML
OpenPepXLLF -ini OPXL2.ini -in path/file2.mzML -out_idXML path/file2.idXML
Setting up and running XFDR (FDR estimation for XL-MS):
Quickstart:
- Generate a default parameter file for XFDR:
XFDR -write_ini XFDR.ini
- Edit parameter file using INIFileEditor (or a text editor) and run XFDR:
XFDR -ini XFDR.ini -in filename.idXML -out_idXML filename.idXML
Full Explanation:
XFDR is a reimplementation of xProphet (Leitner et al., 2014). It reads in cross-link identifications (IDs) and computes score distributions for decoys and targets from the first ranked IDs of each MS2 spectrum. Using these score distributions it assigns an FDR value to every hit. XFDR divides the set if IDs into intra- and inter-protein cross-links as well as mono-links/dead-end-links and loop-links and computes separate score distributions and FDRs for each of these groups. To be able to count the number of intra-protein target-decoy hybrids, XFDR expects that the OpenPepXL/LF search has been done with a corresponding decoy protein for each target protein in the database. The name of each decoy protein should be equal to the target protein with a decoy prefix, e.g. target: “Protein1”, decoy: “decoy_Protein1”. Importantly removing the decoy prefix should make the names exactly the same, in the example case it would be “decoy_”. The specific prefix for decoys can be set to any prefix you wish, but it has to be set consistently in OpenPepXL/LF and XFDR in the -decoy_string parameter.
Some of the parameters of XFDR influence which IDs are used to compute the score distributions for FDR estimation. By default all first ranked IDs are used. The -minborder and -maxborder parameters allow you to set a range of precursor mass errors (the default values of -1 will not apply this filter), e.g. you can compute FDRs from IDs with a precursor mass error between -5 ppm and 5 ppm even if the OpenPepXL/LF search used a precursor tolerance of 10 or 15 ppm. The parameter -mindeltas applies a filter to the score difference between the first and second ranked IDs of an MS2 spectrum. A value of 0.95 will only use IDs with a score ratio of at most 95% between the second and first ranked IDs (or in other words will enforce a score difference of at least 5%). Setting the parameter -uniquexl to “true” will only consider the top scoring ID among those IDs recognized as equal by XFDR. Equal IDs are those that link the same positions in the same digested peptide sequences and are not what one would consider unique cross-links on the protein level.
Setting up and running OpenPepXL (labeled linkers):
Quickstart:
- Generate a default parameter file for FeatureFinderMultiplex (FFM):
FeatureFinderMultiplex -write_ini FFM.ini
- Edit parameter file using INIFileEditor (or a text editor) and run the FFM:
FeatureFinderMultiplex -ini FFM.ini -in filename.mzML -out_multiplets filename.consensusXML
- Generate a default parameter file for OpenPepXL:
OpenPepXL -write_ini OPXL.ini
- Edit parameter file using INIFileEditor (or a text editor) and run the tool using the parameter file:
OpenPepXL -ini OPXL.ini -in filename.mzML -consensus filename.consensusXML -out_idXML filename.idXML
Full Explanation:
All of the parameters as well as the FDR estimation procedure described for OpenPepXLLF above are applicable to OpenPepXL, so this section will mainly describe the additional steps necessary to run OpenPepXL to search for labeled linkers. To use the additional preprocessing features for labeled cross-links, OpenPepXL requires additional information to link together the correct MS2 spectra. This information is provided by a consensusXML file, an OpenMS format containing the boundaries of MS1 features and connections between them. For every .mzML input file, OpenPepXL requires a .consensusXML with linked MS1 features from that .mzML file. Generating a .consensusXML file is a two step process. First finding the MS1 features defined as a group of isotopic mass traces showing a characteristic intensity bell curve along the retention time axis. Each MS1 feature represents one species of molecules, e.g. one peptide or cross-linked peptide pair, localized to a contiguous patch of the two-dimensional m/z vs. retention time map. The second step is linking features at a mass distance that corresponds to the mass difference of the labeled cross-linkers. There are multiple ways of achieving these steps with different TOPP tools. The recommended way is to use FeatureFinderMultiplex. This tool takes care of both steps at once and will only report paired features and discard any unpaired features from the first step.
Setting up FeatureFinderMultiplex (finding feature pairs on the MS1 level):
To run the FeatureFinderMultiplex (FFM) generate an *.ini file (FeatureFinderMultiplex -write_ini FFM.ini) and open it with the INIFileEditor. FFM expects an .mzML input file with centroided MS1 spectra. Choose your .mzML input file and a .consensusXML output file (parameter: -out_multiplets). The parameter value for -algorithm:labels should look like this: [0][mass_difference], e.g. [0][12.07573] for a mixture of labeled DSS linker, DSS-d0 and DSS-d12. Change the -algorithm:charge parameter to what you want to search for (for XL-MS usually 3:7, meaning the range from +3 to +7) and -algorithm:mz_tolerance and -algorithm:mz_unit to your MS instruments MS1 tolerance. This can be set to the same value as the precursor mass tolerance for the OpenPepXL search. Set the parameter -algorithm:rt_band to at least 2, this will make the search more robust and increase the sensitivity by sacrificing a small amount of specificity. Because FFM is used to generate candidate spectrum pairs to search through, sensitivity is more important than specificity. Run FeatureFinderMultiplex in the command line with
FeatureFinderMultiplex -ini FFM.ini
You can now run OpenPepXL using the command line in the same way as OpenPepXLLF with the additional input parameter -consensus filename.consensusXML defined in the INI file or directly as a command line parameter.
Setting up PeakPickerHiRes (centroiding / peak picking of MS1 and/or MS2 spectra):
FeatureFinderMultiplex expects centroided MS1 spectra, while OpenPepXL and OpenPepXLLF expect centroided MS2 spectra. If you have .mzML files that are not centroided or if you are not sure whether they are, you can use PeakPickerHiRes. Generate an INI file (PeakPickerHiRes -write_ini picker.ini) and open it with the INIFileEditor. Choose your input and output file and the MS levels that you want to centroid using the -algorithm:ms_levels parameter. This will force peak picking on these levels, even if they are already picked. You can also leave the -algorithm:ms_levels parameter empty to pick peaks at all MS levels that are not centroided yet. The other settings do not need to be changed from the default for most applications. Run the tool using the command line:
PeakPickerHiRes -ini picker.ini
Visualizing spectra and matched peaks with TOPPView:
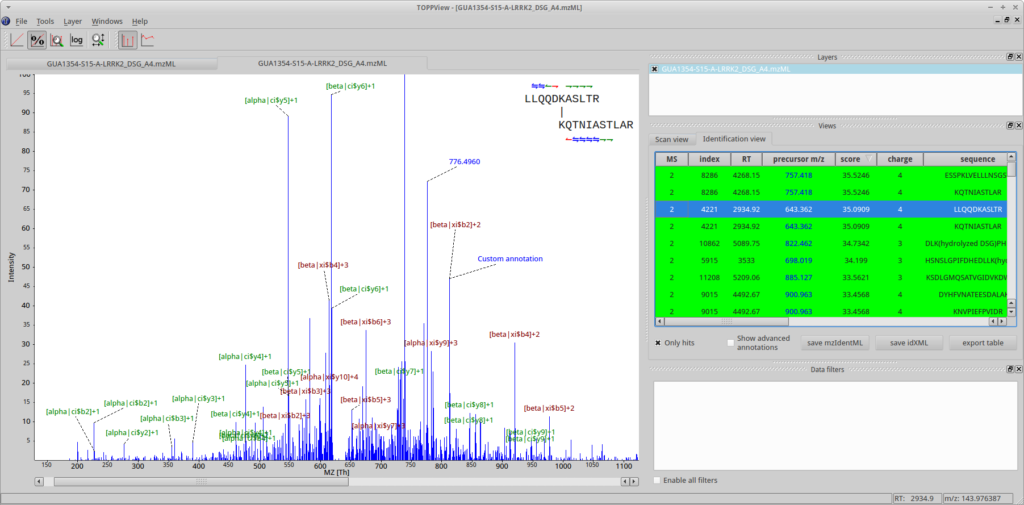
First, open a spectrum in TOPPView. Then go to Tools->Annotate with identification and select the .mzid or .idXML file produced by OpenPepXL. You can select an identified cross-link in the table on the right side of the TOPPView window to visualize it. TOPPView allows you to zoom in and out freely. Peak annotations are read from the identification file but can be edited, moved, added or removed, e.g. to prepare clean images for publication. These custom labels can also be stored to file and retrieved in .idXML files, the internal OpenMS identification data format.
The identified features and feature pairs from FeatureFinderMultiplex (FFM) can also be visualized in TOPPView by loading in the .mzML, then the .featureXML output file (optional, not necessary for OpenPepXL, but contains detailed information about each feature, FFM parameter: -out) and the .consensusXML as additional layers through File->Open file and selecting new layer in the appearing pop-up window.
References
Rinner O, Seebacher J, Walzthoeni T, Mueller L, Beck M, Schmidt A, Mueller M, Aebersold R (2008) Identification of cross-linked peptides from large sequence databases.
Leitner A, Walzthoeni T, Aebersold R. (2014) Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline.
Walzthoeni T, Joachimiak LA, Rosenberger G, Röst HL, Malmström L, Leitner A, Frydman R, Aebersold R (2015) xTract: software for characterizing conformational changes of protein complexes by quantitative cross-linking mass spectrometry.
Kosinski, J., et al. (2015) Xlink Analyzer: Software for analysis and visualization of cross-linking data in the context of three-dimensional structures. J. Struct. Biol.
Vizcaíno, J. A., Mayer, G., Perkins, S. R., Barsnes, H., Vaudel, M., Perez-Riverol, Y., … & Rappsilber, J. (2017). The mzIdentML data standard version 1.2, supporting advances in proteome informatics. Molecular & Cellular Proteomics, mcp-M117.
Ternent, T., Csordas, A., Qi, D., Gómez‐Baena, G., Beynon, R.J., Jones, A.R., Hermjakob, H. and Vizcaíno, J.A., 2014. How to submit MS proteomics data to ProteomeXchange via the PRIDE database. Proteomics, 14(20), pp.2233-2241.