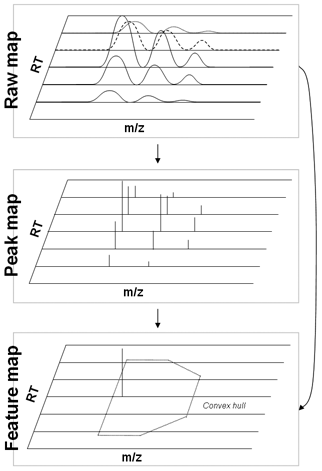
Data reduction in LC-MS analysis mostly consists of two steps. In the first step, called "peak picking", important information of the mass spectrometric peaks (e.g. peaks' mass centroid positions, their areas under curve and full-width-at-half-maxima) are extracted from the raw LC-MS data. The second data reduction step, called "feature finding", represents the quantitation of all peptides in a proteomic sample. Therefore, the signals in a LC-MS map caused by all charge and isotopic variants of the peptide are detected and summarized resulting in a list of compounds or features, each characterized by mass, retention time and abundance. The classes described in this section can be found in the TRANSFORMATIONS folder.
Peak picking
For peak picking, the class PeakPickerCWT or PeakPickerHiRes is used. Because this class detects and extracts mass spectrometric peaks it is applicable to LC-MS as well as MALDI raw data.
The following example (Tutorial_PeakPickerCWT.cpp) shows how to open a raw map, initialize a PeakPickerCWT object, set the most important parameters (the scale of the wavelet, a peak's minimal height and FWHM), and start the peak picking process.
The output of the program is:
- Note
- A rough standard value for the peak's scale is the average FWHM of a mass spectrometric peak.
Feature detection
The FeatureFinders implement different algorithms for the detection and quantitation of peptides from LC-MS maps. In contrast to the previous step (peak picking), we do not only search for pronounced signals (peak) in the LC-MS map but search explicitly for peptides which can be recognized by their isotopic pattern.
OpenMS offers different algorithms for this task. Details of how to apply them are given in the TOPP documentation. Please also refer to our publications on the OpenMS web page. TOPP contains multiple command line programs which allow to execute our algorithms without writing a single line of code.
But you can also write your own FeatureFinder application. This gives you more flexibility and is straightforward to do. A short example (Tutorial_FeatureFinder.cpp) is given below. First we need to instantiate the FeatureFinder, its parameters and the input/output data:
Then we run the FeatureFinder. The first argument is the algorithm name (here 'simple'). Using the second and third parameter, the peak and feature data is handed to the algorithm. The fourth argument sets the parameters used by the algorithm.