|
OpenMS
2.5.0
|
This class implements a pair finding algorithm for consensus features. More...
#include <OpenMS/ANALYSIS/MAPMATCHING/StablePairFinder.h>
Public Types | |
| typedef BaseGroupFinder | Base |
| Base class. More... | |
 Public Types inherited from ProgressLogger Public Types inherited from ProgressLogger | |
| enum | LogType { CMD, GUI, NONE } |
| Possible log types. More... | |
Public Member Functions | |
| StablePairFinder () | |
| Constructor. More... | |
| ~StablePairFinder () override | |
| Destructor. More... | |
| void | run (const std::vector< ConsensusMap > &input_maps, ConsensusMap &result_map) override |
| Run the algorithm. More... | |
| Public Member Functions inherited from BaseGroupFinder | |
| BaseGroupFinder () | |
| Default constructor. More... | |
| ~BaseGroupFinder () override | |
| Destructor. More... | |
| Public Member Functions inherited from DefaultParamHandler | |
| DefaultParamHandler (const String &name) | |
| Constructor with name that is displayed in error messages. More... | |
| DefaultParamHandler (const DefaultParamHandler &rhs) | |
| Copy constructor. More... | |
| virtual | ~DefaultParamHandler () |
| Destructor. More... | |
| virtual DefaultParamHandler & | operator= (const DefaultParamHandler &rhs) |
| Assignment operator. More... | |
| virtual bool | operator== (const DefaultParamHandler &rhs) const |
| Equality operator. More... | |
| void | setParameters (const Param ¶m) |
| Sets the parameters. More... | |
| const Param & | getParameters () const |
| Non-mutable access to the parameters. More... | |
| const Param & | getDefaults () const |
| Non-mutable access to the default parameters. More... | |
| const String & | getName () const |
| Non-mutable access to the name. More... | |
| void | setName (const String &name) |
| Mutable access to the name. More... | |
| const std::vector< String > & | getSubsections () const |
| Non-mutable access to the registered subsections. More... | |
| Public Member Functions inherited from ProgressLogger | |
| ProgressLogger () | |
| Constructor. More... | |
| ~ProgressLogger () | |
| Destructor. More... | |
| ProgressLogger (const ProgressLogger &other) | |
| Copy constructor. More... | |
| ProgressLogger & | operator= (const ProgressLogger &other) |
| Assignment Operator. More... | |
| void | setLogType (LogType type) const |
| Sets the progress log that should be used. The default type is NONE! More... | |
| LogType | getLogType () const |
| Returns the type of progress log being used. More... | |
| void | startProgress (SignedSize begin, SignedSize end, const String &label) const |
| Initializes the progress display. More... | |
| void | setProgress (SignedSize value) const |
| Sets the current progress. More... | |
| void | endProgress () const |
| Ends the progress display. More... | |
| void | nextProgress () const |
| increment progress by 1 (according to range begin-end) More... | |
Static Public Member Functions | |
| static BaseGroupFinder * | create () |
| Returns an instance of this class. More... | |
| static const String | getProductName () |
| Returns the name of this module. More... | |
| Static Public Member Functions inherited from BaseGroupFinder | |
| static void | registerChildren () |
| Register all derived classes here. More... | |
Internal helper classes and enums | |
| enum | { RT = Peak2D::RT, MZ = Peak2D::MZ } |
| double | second_nearest_gap_ |
| The distance to the second nearest neighbors must be by this factor larger than the distance to the matched element itself. More... | |
| bool | use_IDs_ |
| Only match if peptide IDs are compatible? More... | |
| void | updateMembers_ () override |
| This method is used to update extra member variables at the end of the setParameters() method. More... | |
| bool | compatibleIDs_ (const ConsensusFeature &feat1, const ConsensusFeature &feat2) const |
| Checks if the peptide IDs of two features are compatible. More... | |
| const AASequence & | getBestHitSequence_ (const PeptideIdentification &peptideIdentification) const |
| Returns the highest scoring peptide hit in the the given peptide identification. More... | |
Additional Inherited Members | |
| Protected Member Functions inherited from BaseGroupFinder | |
| void | checkIds_ (const std::vector< ConsensusMap > &maps) const |
| Checks if all file descriptions have disjoint map identifiers. More... | |
| Protected Member Functions inherited from DefaultParamHandler | |
| void | defaultsToParam_ () |
| Updates the parameters after the defaults have been set in the constructor. More... | |
| Static Protected Member Functions inherited from ProgressLogger | |
| static String | logTypeToFactoryName_ (LogType type) |
| Return the name of the factory product used for this log type. More... | |
| Protected Attributes inherited from DefaultParamHandler | |
| Param | param_ |
| Container for current parameters. More... | |
| Param | defaults_ |
| Container for default parameters. This member should be filled in the constructor of derived classes! More... | |
| std::vector< String > | subsections_ |
| Container for registered subsections. This member should be filled in the constructor of derived classes! More... | |
| String | error_name_ |
| Name that is displayed in error messages during the parameter checking. More... | |
| bool | check_defaults_ |
| If this member is set to false no checking if parameters in done;. More... | |
| bool | warn_empty_defaults_ |
| If this member is set to false no warning is emitted when defaults are empty;. More... | |
| Protected Attributes inherited from ProgressLogger | |
| LogType | type_ |
| time_t | last_invoke_ |
| ProgressLoggerImpl * | current_logger_ |
| Static Protected Attributes inherited from ProgressLogger | |
| static int | recursion_depth_ |
This class implements a pair finding algorithm for consensus features.
It offers a method to determine pairs across two consensus maps. The corresponding consensus features must be aligned, but may have small position deviations.
The distance measure is implemented in class FeatureDistance - see there for details.
Additional criteria for pairing
Depending on parameter use_identifications, peptide identifications annotated to the features may have to be compatible (i.e. no annotation or the same annotation) for a pairing to occur.
Stability criterion: The distance to the nearest neighbor must be smaller than the distance to the second-nearest neighbor by a certain factor, see parameter second_nearest_gap. There is a non-trivial relation between this parameter and the maximum allowed difference (in RT or m/z) of the distance measure: If second_nearest_gap is greater than one, lowering max_difference may in fact lead to more - rather than fewer - pairings, because it increases the distance difference between the nearest and the second-nearest neighbor, so that the constraint imposed by second_nearest_gap may be fulfilled more often.
Quality calculation
The quality of a pairing is computed from the distance between the paired elements (nearest neighbors) and the distances to the second-nearest neighbors of both elements, according to the formula:
![\[ q_{i,j} = \big( 1 - d_{i,j} \big) \cdot \big( 1 - \frac{g \cdot d_{i,j}}{d_{2,i}} \big) \cdot \big( 1 - \frac{g \cdot d_{i,j}}{d_{2,j}} \big) \cdot \]](form_18.png)
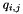 is the quality of the pairing of elements i and j,
is the quality of the pairing of elements i and j, 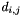 is the distance between the two,
is the distance between the two, 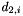 and
and 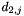 are the distances to the second-nearest neighbors of i and j, respectively, and g is the factor defined by parameter
are the distances to the second-nearest neighbors of i and j, respectively, and g is the factor defined by parameter second_nearest_gap.
Note that by the definition of the distance measure, 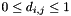 if i and j are to form a pair. The criteria for pairing further require that
if i and j are to form a pair. The criteria for pairing further require that 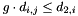 and
and 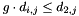 . This ensures that the resulting quality is always between one (best) and zero (worst).
. This ensures that the resulting quality is always between one (best) and zero (worst).
For the final quality q of the consensus feature produced by merging two paired elements (i and j), the existing quality values of the two elements are taken into account. The final quality is a weighted average of the existing qualities ( 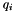 and
and 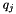 ) and the quality of the pairing ( , see above):
) and the quality of the pairing ( , see above):
![\[ q = \frac{q_{i,j} + (s_i - 1) \cdot q_i + (s_j - 1) \cdot q_j}{s_i + s_j - 1} \]](form_28.png)
The weighting factors 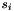 and
and 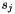 are the sizes (i.e. numbers of subelements) of the two consensus features i and j. That way, it is possible to link several feature maps to a growing consensus map in a stepwise fashion (as done by FeatureGroupingAlgorithmUnlabeled), and in the end obtain quality values that incorporate the qualities of all pairings that occurred during the generation of a consensus feature. Note that "missing" elements (if a consensus feature does not contain sub-features from all input maps) are not punished in this definition of quality.
are the sizes (i.e. numbers of subelements) of the two consensus features i and j. That way, it is possible to link several feature maps to a growing consensus map in a stepwise fashion (as done by FeatureGroupingAlgorithmUnlabeled), and in the end obtain quality values that incorporate the qualities of all pairings that occurred during the generation of a consensus feature. Note that "missing" elements (if a consensus feature does not contain sub-features from all input maps) are not punished in this definition of quality.
| Name | Type | Default | Restrictions | Description |
|---|---|---|---|---|
| second_nearest_gap | float | 2.0 | min: 1.0 | Only link features whose distance to the second nearest neighbors (for both sides) is larger by 'second_nearest_gap' than the distance between the matched pair itself. |
| use_identifications | string | false | true, false | Never link features that are annotated with different peptides (features without ID's always match; only the best hit per peptide identification is considered). |
| ignore_charge | string | false | true, false | false [default]: pairing requires equal charge state (or at least one unknown charge '0'); true: Pairing irrespective of charge state |
| ignore_adduct | string | true | true, false | true [default]: pairing requires equal adducts (or at least one without adduct annotation); true: Pairing irrespective of adducts |
| distance_RT:max_difference | float | 100.0 | min: 0.0 | Never pair features with a larger RT distance (in seconds). |
| distance_RT:exponent | float | 1.0 | min: 0.0 | Normalized RT differences ([0-1], relative to 'max_difference') are raised to this power (using 1 or 2 will be fast, everything else is REALLY slow) |
| distance_RT:weight | float | 1.0 | min: 0.0 | Final RT distances are weighted by this factor |
| distance_MZ:max_difference | float | 0.3 | min: 0.0 | Never pair features with larger m/z distance (unit defined by 'unit') |
| distance_MZ:unit | string | Da | Da, ppm | Unit of the 'max_difference' parameter |
| distance_MZ:exponent | float | 2.0 | min: 0.0 | Normalized ([0-1], relative to 'max_difference') m/z differences are raised to this power (using 1 or 2 will be fast, everything else is REALLY slow) |
| distance_MZ:weight | float | 1.0 | min: 0.0 | Final m/z distances are weighted by this factor |
| distance_intensity:exponent | float | 1.0 | min: 0.0 | Differences in relative intensity ([0-1]) are raised to this power (using 1 or 2 will be fast, everything else is REALLY slow) |
| distance_intensity:weight | float | 0.0 | min: 0.0 | Final intensity distances are weighted by this factor |
| distance_intensity:log_transform | string | disabled | enabled, disabled | Log-transform intensities? If disabled, d = |int_f2 - int_f1| / int_max. If enabled, d = |log(int_f2 + 1) - log(int_f1 + 1)| / log(int_max + 1)) |
| typedef BaseGroupFinder Base |
Base class.
| StablePairFinder | ( | ) |
Constructor.
|
inlineoverride |
Destructor.
|
protected |
Checks if the peptide IDs of two features are compatible.
A feature without identification is always compatible. Otherwise, two features are compatible if the best peptide hits of their identifications have the same sequences.
|
inlinestatic |
Returns an instance of this class.
|
protected |
Returns the highest scoring peptide hit in the the given peptide identification.
| peptideIdentification | The peptideIdentification to scan. |
|
inlinestatic |
Returns the name of this module.
|
overridevirtual |
Run the algorithm.
| Exception::IllegalArgument | is thrown if the input data is not valid. |
Implements BaseGroupFinder.
|
overrideprotectedvirtual |
This method is used to update extra member variables at the end of the setParameters() method.
Also call it at the end of the derived classes' copy constructor and assignment operator.
The default implementation is empty.
Reimplemented from DefaultParamHandler.
|
protected |
The distance to the second nearest neighbors must be by this factor larger than the distance to the matched element itself.
|
protected |
Only match if peptide IDs are compatible?
 1.8.16
1.8.16

