OpenMS is a flexible codebase that can be tailored to many different applications ranging from the standard label free analysis to top down, metabolomics, crosslinking or DIA.
Choose an application of OpenMS that you are interested in. The pages will provide explanations on how OpenMS can be used to solve your problems and link to workflows that allow you to apply the tools to your data.
If you cannot find your application in the menu on the left, more OpenMS tools can be found in the TOPP documentation.
DIAMetAlyzer is a KNIME workflow which integrates DDA and targeted DIA analysis for metabolomics, which allows for false-discovery rate estimation based a target-decoy approach (see figure 1). It performs DDA based candidate identification and constructs a target/decoy assay library, which is used for DIA target extraction and statistical validation (FDR estimation)
Fig. 1: DIAMetAlyzer – pipeline for assay library generation and targeted analysis with statistical validation
Candidate identification includes feature detection, adduct grouping and accurate mass search.
DIAproteomics is a bioinformatics analysis pipeline used for quantitative processing of data independent (DIA) proteomics data.
The workflow is based on the OpenSwathWorkflow for SWATH-MS proteomic data. DIA RAW files (mzML) serve as inputs and library search is performed based on a given input spectral library. If specified internal retention time standards (irts) will be used to align library and DIA measurements into the same retention time space. FDR rescoring is applied using Pyprophet based on a competitive target-decoy approach on peakgroup or global peptide and protein level.
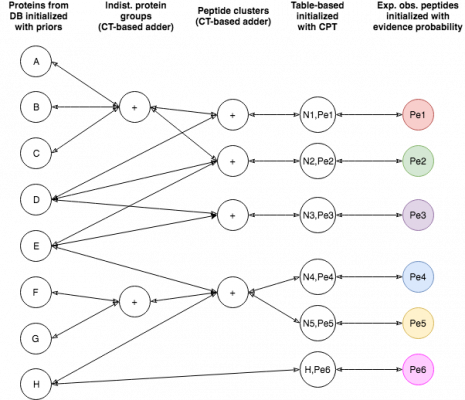
EPIFANY is a tool for efficient bayesian protein inference. It is included in OpenMS since 2.5.
It takes one or more peptide database search engine results (in OpenMS' idXML format) that were post-processed
by the PercolatorAdapter or IDPosteriorErrorProbability tool and adds posterior probabilities and/or
false discovery rates for each protein or protein group to it.
Ultra-fast MS1/MS2 deconvolution for top-down proteomics
FLASHDECONV 2.0 BETA+, FINALLY WITH A GUI! Finally a GUI is here. You can find the GUI command in [OpenMS path]/bin folder. Go to [OpenMS path]/bin and run FLASHDeconvWizard! FLASHDeconv 2.0 beta+ works for MS1 and MS2 spectral deconvolution and feature deconvolution. It supports various output formats (e.g., *.tsv, *.mzML, *.msalign, and *.feature). FLASHDeconv 2.0 stable version will be officially integrated in OpenMS 2.7.0 released in near future. FLASHDeconv 2.0 beta+ also supports TopPIC identification better than the previous version, by generating all msalign and feature files for TopPIC inputs.
Intelligent data acquisition for top-down proteomics
FLASHIda FLASHIda is an intelligent online data acquisition algorithm for top-down proteomics (TDP) that ensures the real-time selection of high-quality precursors of diverse proteoforms. FLASHIda combines fast decharging algorithms in FLASHDeconv and machine learning-based quality assessment to identify optimal precursors for fragmentation. Currently the C# source code and instruction of FLASHIda is available in here under a BSD three-clause license. We are working on merging FLASHIda into OpenMS.
Info FLASHIda beta version (under construction)
FLASHQuant performs MS1-level label-free quantification data analysis in top-down proteomics with an automatic overlapping signal resolution method.
Check it out on Github! Now binary installer files for all platforms are available here: (Please use the latest version) Download Page
Input/Output Input: Centroided MS1 scans (*.mzML) Output: Quantified proteoforms in a tab-separated file (*.tsv); optionally, OpenMS LC-MS features output (*.featureXML) for each proteoform, mono-isotopic/average mass, retention time range, charge range, different types for quantity values, and isotope cosine similarity score are provided.
MetaProSIP: automated inference of elemental fluxes in microbial communities Info MetaProSIP has been fully integrated into OpenMS. No additional installer is required anymore. Update: MetaProSIP now supports labeling experiments with heavy water (D, 18O)
Download an updated KNIME workflow: here
Advanced workflow including mass recalibration, alignment, and ID pooling: here
Requirements High-Resolution MS, CID, or HCD MS2 Developed and tested on orbitrap instruments MS1 and MS2 need to be centroided (either on acquisition, conversion or in a workflow using the TOPP tool HiResPeakPicker) For additional information on sample handling and acquisition please refer to the original MetaProSIP publication.
Quantification and identification workflow for MHC peptides
MHCquant: Identify and quantify peptides from mass spectrometry raw data
MHCquant is an analysis pipeline used for quantitative processing of data dependent (DDA) peptidomics data.
It was specifically designed to analyze immunopeptidomics data, which deals with the analysis of affinity-purified, unspecifically cleaved peptides that have recently been discussed intensively in the context of cancer vaccines. (https://www.nature.com/articles/ncomms13404)
The workflow is based on the OpenMS C++ framework for computational mass spectrometry.
NASE is now included in OpenMS release, 2.5.
Requirements:
HCD (or ETD) data of RNA oligonucleotides acquired on a high-resolution mass spectrometer Fragment spectra (MS/MS) need to be centroided (either on acquisition, conversion, or in a workflow using the TOPP tool PeakPickerHiRes) Developed and tested on Linux (Ubuntu 18.04 and 18.10) systems with data from orbitrap instruments Operating system: OpenMS installers have been tested on Ubuntu Linux 18.04, Windows 7/8/10, and macOS 10.
Introduction NuXL is a novel tool for protein-RNA and DNA cross-linking studies. It is available as a stand-alone tool and as a Proteome Discoverer community node or as a web application that doesn’t require you to install additional software. This guide outlines the steps to install NuXL, set up your analysis, and interpret your data using the provided tools and workflows.
Requirements: HCD data acquired on a high-resolution MS Developed and tested on orbitrap instruments (including Velos, Lumos, and Astral) Installation for Proteome Discoverer NuXL is currently compatible with Proteome Discoverer 3.
OpenPepXL: an open source peptide cross-link identification tool OpenPepXL is a protein-protein cross-link identification tool implemented in C++ as part of OpenMS. It works with all uncleavable labeled and label-free cross-linkers but not (yet) with cleavable ones.
Requirements The current version of OpenPepXL, version 1.1 is available as part of OpenMS 2.5. Installers for Windows, MacOS and Linux can be found here. OpenPepXL can be used effectively on a desktop computer with 16BG of memory.